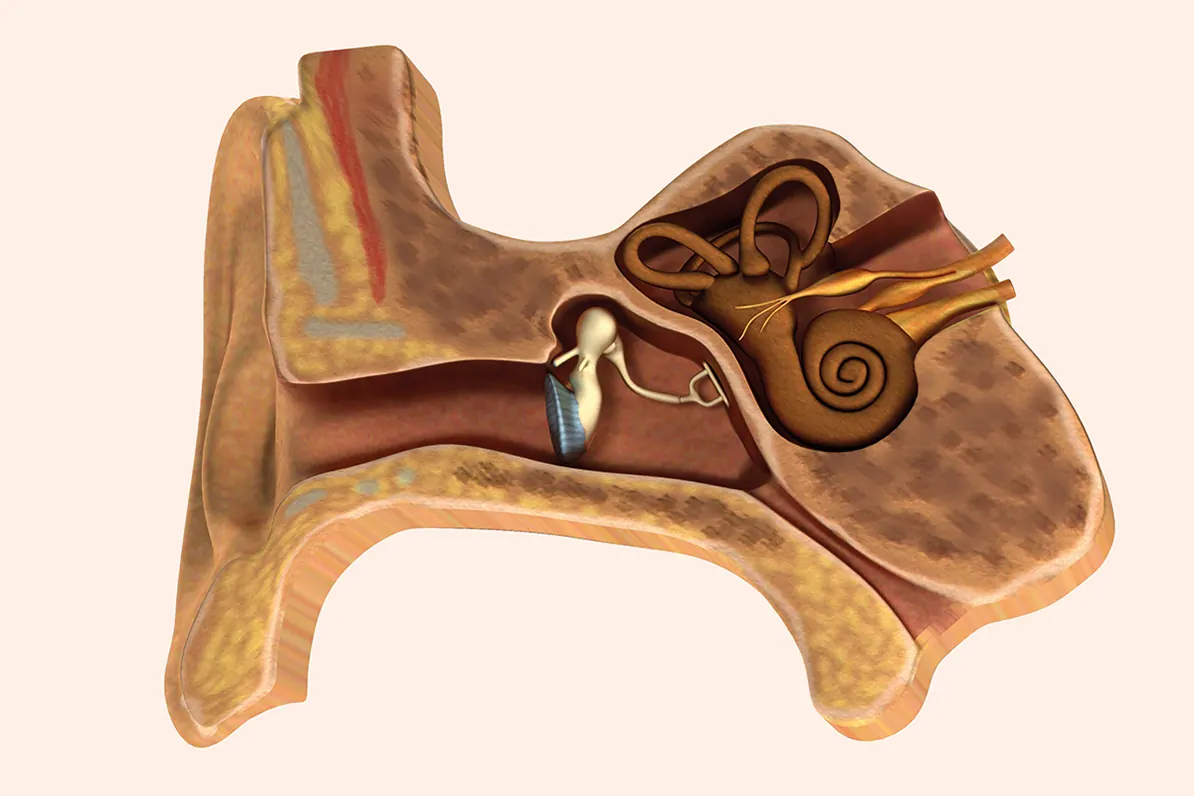
La pérdida de audición es el déficit sensorial más prevalente en la biología
humana, afectando actualmente a más de 1.500 millones de personas en todo
el mundo, incluyendo todas las formas y grados de hipoacusia. De esta cifra,
aproximadamente 430 millones presentan una pérdida auditiva discapacitante,
una crisis de salud pública que afecta a 34 millones de niños. En el ámbito
pediátrico, el impacto es especialmente crítico, ya que el 60% de los casos
de sordera congénita tiene una raíz genética documentada.
Durante más de un siglo, la medicina y la ciencia se centraron en la compensación de la pérdida auditiva mediante dispositivos mecánicos como audífonos e implantes cocleares. De hecho, en 1972 se realizó el primer implante coclear monocanal documentado y durante las décadas posteriores, especialmente a partir de la década de los 80, el desarrollo de los implantes multicanal supuso un hito en el tratamiento de la hipoacusia profunda, transformando la rehabilitación auditiva en niños y en adultos, en todo el mundo.
Paralelamente, en los primeros años 70 emergió la terapia génica en el ámbito experimental, si bien el primer ensayo clínico exitoso en humanos data de 1990. En aquel momento el tratamiento no consiguió una cura completa (se trataba de una inmunodeficiencia grave), pero fue un paso clave que abrió la puerta a un gran número de investigaciones posteriores para otras enfermedades. Desde entonces, las terapias génicas han avanzado de forma exponencial y, de hecho, en la última década ya se aplica en tratamientos aprobados por la FDA y la EMA para enfermedades hereditarias de la retina, trastornos metabólicos raros y algunos tipos de cáncer.
A partir de la década de los 80, el desarrollo de los implantes multicanal supuso un hito en el tratamiento de la hipoacusia profunda, transformando la rehabilitación auditiva en niños y en adultos.
En lo relativo a la restauración biológica de la audición mediante la terapia génica, el año 2024 marca un punto de inflexión histórico, pero para comprender la magnitud de los avances actuales, es imperativo analizar la evolución del pensamiento científico desde sus inicios.
Toda esta revolución se remonta al siglo XIX, cuando la biología vegetal adoptó conocimientos agrícolas para estructurar la genética mendeliana. Durante este siglo, los bioquímicos caracterizaron por primera vez el núcleo celular y los ácidos nucleicos, estableciendo la noción de que los rasgos se heredan a través de partículas específicas. En aquella época, se creía que la sangre era el mecanismo de herencia, pero los experimentos botánicos demostraron que existía un manual de instrucciones molecular.
El primer ensayo clínico con terapia génica exitoso en humanos data de 1990 y en la última década, ya se aplica en tratamientos aprobados por la FDA y la EMA para enfermedades hereditarias de la retina, trastornos metabólicos raros y algunos tipos de cáncer.
Esta evolución continuó en la década de los 50 del pasado siglo con el descubrimiento del «principio transformador» que demostró que el ADN era la sustancia física de la herencia y que por tanto, quizá sería posible introducir o corregir un gen en las células humanas para tratar una enfermedad desde su causa molecular.

Así, experimentos con bacteriófagos (virus cuya propiedad es que infectan solo a bacterias), revelaron que los virus podían actuar como vectores, transportando material genético entre organismos sin transferir proteínas virales. Este hallazgo constituye la piedra angular de la terapia génica moderna: si un virus puede inyectar su código en una célula, es posible «secuestrar» esa capacidad para introducir genes sanos que reemplacen a los defectuosos.
El descubrimiento del «principio transformador», en los años 50, demostró que el ADN era la sustancia física de la herencia, lo que hacía posible tratar una enfermedad desde su causa molecular introduciendo o corrigiendo un gen en las células humanas.
No obstante, esta visión técnica ha favorecido la teoría del «determinismo genético», la creencia de que manipular un solo gen es la solución definitiva para fenotipos tan complejos como la audición humana, partiendo de la idea de que nuestro ADN define nuestro destino biológico en su totalidad.
Hoy en día parece claro que el determinismo genético absoluto no existe y que hay factores coadyuvantes que deben tomarse en consideración, como la interacción genes-ambiente, o los llamados genes de «susceptibilidad», que, como la propia palabra indica, aumentan la probabilidad de desarrollar la enfermedad, pero no lo garantizan.
Como se ha comentado previamente, en lo que a terapia génica de la hipoacusia se refiere, el año 2024 ha marcado un punto crítico de inflexión, ya que durante este año se reportaron los resultados de los primeros ensayos clínicos de terapia génica en personas con hipoacusia hereditaria (concretamente en la originada por mutaciones del gen OTOF, responsable de producir una proteína llamada otoferlina, crítica para la liberación de neurotransmisores desde las células ciliadas del oído interno).
Estos primeros ensayos mostraron mejoras auditivas reales tras una sola administración, un hallazgo sin precedentes en la historia del tratamiento de la hipoacusia. En el ensayo clínico DB-OTO, de Regeneron, en el que ha participado España desde la aprobación del ensayo clínico por parte de la Agencia Española de Medicamentos y Productos Sanitarios en mayo de 2023, se documentaron mejoras auditivas significativas e incluso normalización de los umbrales auditivos en algunos casos después de 24 semanas. El reclutamiento en nuestro país se inició según las fuentes consultadas en marzo de 2024.
El determinismo genético absoluto no existe, hay factores coadyuvantes que deben tomarse en consideración, como la interacción genes-ambiente o los llamados genes de «susceptibilidad».

El estudio se encuentra en Fase ½, esto es, en proceso de evaluación de su seguridad y tolerabilidad y de documentación de señales preliminares de eficacia, y aparentemente continúa activo en este momento, si bien la administración del tratamiento depende de criterios muy específicos de los centros autorizados.
El avance más tangible reportado recientemente es el éxito del primer ensayo clínico de terapia génica bilateral en niños con sordera recesiva tipo 9 (DFNB9), una mutación en el gen OTOF mencionado previamente.
En el ensayo clínico DB-OTO, de Regeneron, se documentaron mejoras auditivas significativas e incluso normalización de los umbrales auditivos en algunos casos después de 24 semanas.
En el estudio, publicado en Nature Medicine, los investigadores administraron el vector viral AAV1-hOTOF en ambos oídos de cinco pacientes pediátricos (de entre 1 y 11 años) mediante una sola intervención quirúrgica. Los resultados de este ensayo clínico han sido calificados como transformadores para la otología y la audiología actual. Entre ellos, cabe destacar los siguientes:
1. Restauración de umbrales: pacientes que inicialmente no respondían a sonidos por debajo de los 95 dB, recuperaron la audición bilateral con umbrales que bajaron hasta rangos de 50 a 58 dB en apenas 26 semanas.
2. Localización del sonido: a diferencia de la terapia unilateral, la administración binaural permitió que los niños recuperaran la capacidad de localizar la fuente del sonido. Las pruebas mostraron que el error cuadrático medio (RMSE) de localización sonora mejoró de 92.8° a 40.0°.
3. Hitos del desarrollo: el estudio documentó cómo los niños comenzaron a pronunciar sílabas y palabras sencillas como «baba» (padre) y «mama» (madre), y mostraron la capacidad de bailar al ritmo de la música por primera vez.
Este ensayo demostró que la administración bilateral en una sola cirugía es segura y eficiente, evitando el riesgo de que el sistema inmune genere anticuerpos neutralizantes tras una primera dosis, lo que podría impedir un tratamiento posterior en el segundo oído.
Pero la terapia génica plantea otros retos. Así, por ejemplo, parece ser que el tamaño de los genes auditivos constituye un obstáculo técnico importante. Para explicarlo de la forma más sencilla posible, el vector de virus adenoasociado (AAV), comúnmente utilizado por su baja patogenicidad, tiene una capacidad de carga limitada a 4,7 kilobases (kb). Sin embargo, el gen OTOF humano mide cerca de 6 kb, lo que supera este límite físico. Para solucionar esto, los científicos han diseñado el sistema de vector AAV dual. En este enfoque, el gen terapéutico se corta en dos mitades (segmentos 5’ y 3’), se empaquetan en dos vectores distintos y, una vez inyectados en el oído, ambas mitades deben infectar la misma célula simultáneamente. Dentro de la célula ciliada, los fragmentos se ensamblan para reconstituir la proteína funcional completa. Aunque ingeniosa, esta técnica plantea retos de eficiencia, ya que requiere una carga viral mayor para asegurar que ambos componentes coincidan en la misma célula, lo que incrementa el riesgo de inmunotoxicidad.

Otra línea actual de investigación se centra en explorar estrategias para tratar la pérdida auditiva adquirida, como la inducida por ruido o por fármacos ototóxicos como el cisplatino, utilizado en quimioterapia.
El avance reciente más tangible, es el éxito del primer ensayo clínico de terapia génica bilateral en niños con sordera recesiva tipo 9 (DFNB9), una mutación del gen OTOF.
En estos casos, el objetivo no es siempre el reemplazo génico, sino la entrega de neurotrofinas, proteínas que actúan como factores de supervivencia. Protegen las neuronas del ganglio espiral y las sinapsis de cinta (ribbonsynapses en inglés), que son las primeras estructuras en degenerarse tras un trauma acústico. Estudios preclínicos han demostrado que la sobreexpresión de neurotrofinasNT3 puede regenerar las conexiones perdidas, restaurando la comunicación entre el oído y el cerebro. Incluso se investigan factores de transcripción como ATOH1, clave para la regeneración de células ciliadas, aunque los ensayos en humanos han mostrado que su éxito depende de otros factores, como la reinnervación nerviosa simultánea.
Algunos estudios han centrado su atención en mejorar la precisión de la cirugía y las rutas de administración, ya que de ello depende en gran medida el éxito de la terapia. Las fuentes consultadas describen cuatro rutas principales:
• Membrana de la Ventana Redonda (RWM): es la vía más estudiada en humanos; el vector se inyecta directamente a través de esta membrana hacia el fluido perilinfático.
• Canalostomía: inyección a través de los canales semicirculares, diseñada para minimizar el trauma iatrogénico en pacientes que conservan audición residual.
• Cocleostomía: acceso directo a la rampa media para bañar las células ciliadas en endolinfa, aunque conlleva un mayor riesgo de pérdida de potencial endococlear e inflamación.
• Entrega vía Líquido Cefalorraquídeo (LCR): una técnica emergente que aprovecha la comunicación entre el cerebro y el oído a través del acueducto coclear, evitando la cirugía invasiva del temporal, pero con riesgo de distribución sistémica del gen.

Tomando en consideración la velocidad de estos avances, un panel multidisciplinar de 46 expertos mundiales de China, EE. UU., Reino Unido, Alemania y otros países, ha establecido el primer Consenso Internacional sobre Terapia Génica para la Pérdida Auditiva Hereditaria. Este marco es vital para garantizar la seguridad del paciente y estandarizar los ensayos clínicos.
Los puntos fundamentales del consenso incluyen:
1. Edad de Intervención: se establece una edad mínima recomendada de 10 meses, priorizando la recuperación temprana para maximizar la ventana de plasticidad cerebral necesaria para el lenguaje.
2. Criterios de Selección Preferente: individuos con pérdida auditiva de severa a profunda con variantes patogénicas confirmadas mediante secuenciación de nueva generación.
3. Evaluación Preoperatoria: se establece como obligatorio el uso de Resonancia Magnética (MRI) y Tomografía de Alta Resolución (HRCT) para descartar malformaciones estructurales.
4. Seguimiento y Seguridad: se exige un monitoreo postoperatorio de al menos 5 años, e idealmente de por vida, para detectar efectos adversos tardíos como la oncogénesis o respuestas inmunes severas.
5. Protocolo Farmacológico: se recomienda el uso de glucocorticoides (como dexametasona) para mitigar la respuesta inflamatoria contra el vector viral.
No obstante, y a pesar del entusiasmo mediático, algunas voces académicas como las de O’Neil Guthrie, en su reciente publicación de 2025 en la revista International Journal of Molecular Sciences, instan a la prudencia y a un rigor científico extremo, y advierten sobre el fenómeno del sensacionalismo. Guthrie señala que los fracasos clínicos (como el del gen ATOH1, que fue un fracaso parcial, porque tampoco hubo eventos adversos graves) a menudo se entierran en el silencio informativo, creando una falsa percepción de éxito garantizado.

Desde el punto de vista biológico, parece que el riesgo más grave es la mutagénesis insertacional. Esto ocurre cuando el ADN viral se integra de forma errónea en el genoma del paciente, activando potencialmente oncogenes que pueden provocar cáncer años después del tratamiento, o inactivando genes supresores de tumores. Además, la inyección de dosis altas de vectores puede desencadenar «tormentas de citoquinas», respuestas inmunes masivas y descontroladas que ya han causado muertes en otros campos de la medicina genética.
El «milagro de la audición biológica» es casi una realidad desde el punto de vista técnico, pero según los expertos, para garantizar su seguridad, el futuro debe construirse sobre la transparencia científica y la vigilancia biológica a largo plazo.
Así las cosas, los investigadores clínicos se enfrentan a un profundo dilema ético. La cuestión clave sobre la mesa es si es razonable para la ciencia asumir riesgos de mortalidad por una condición que no pone en riesgo la vida, como es la pérdida auditiva. De hecho, en la comunidad médica se expande el temor de que las familias de los niños con hipoacusia severa o profunda rechacen el implante coclear, tecnología por otra parte sobradamente probada, en aras de una cura génica que podría llegar demasiado tarde para el desarrollo del habla.
El «milagro de la audición biológica» es hoy día casi una realidad desde el punto de vista técnico. Pero, según la opinión de los expertos, para garantizar su seguridad, el futuro debe construirse sobre la transparencia científica y la vigilancia biológica a largo plazo, no sobre el entusiasmo comercial. No debe verse todavía como un reemplazo absoluto de las herramientas actuales, sino como un complemento potente en un enfoque de salud auditiva personalizado. La vuelta «biológica» al sonido debe ser, por encima de todo, una intervención honesta y segura para todos.
Bibliografía:
Duhon, B. H., Bielefeld, E. C., Ren, Y., & Naidoo, J. (2024). Gene therapy advancements for the treatment of acquired and hereditary hearing loss. Frontiers in Audiology and Otology, 2:1423853. https://doi.org/10.3389/fauot.2024.1423853.
Fan, X., Gao, Z., Zhong, J., Chen, Y., Chen, X., Landegger, L. D.,. & Shu, Y. (2026). International expert consensus on gene therapy for hereditary hearing loss: Based on clinical trials. Med, 7(100886). https://doi.org/10.1016/j.medj.2025.100886.
Guthrie, O. W. (2025). Gene therapy: An historical overview for familial hearing loss. International Journal of Molecular Sciences, 26(1469). https://doi.org/10.3390/ijms26041469.
Valayannopoulos, V., Bance, M., Herman, G.A., Baras, A., & Whitton, J.P. (2025). DB-OTO gene therapy for inherited deafness. The New England Journal of Medicine. https://doi.org/10.1056/NEJMoa2400521.
Wang, H., Chen, Y., Lv, J., Cheng, X., Cao, Q., Wang, D., … & Shu, Y. (2024). Bilateral gene therapy in children with autosomal recessive deafness 9: Single-arm trial results. Nature Medicine, 30, 1898-1904. https://doi.org/10.1038/s41591-024-03023-5.
CV Autor
Audióloga / Audioprotesista
Licenciada en Pedagogía y Máster de Logopedia.
Técnico Superior en Audiología Protésica.
Especializada en Audiología Infantil y Evaluación de los trastornos del PAC en RV Alfa Centros Auditivos.
Docente en el Máster de Audiología de la Universidad Europea Miguel de Cervantes.